
چند تعریف که در درک بهتر مقالات چاپ شده در مورد تعیین نوع آمینو اسید از روی شیفت شیمیایی در
NMR
کمک میکنند:
What is the E-Value in BLAST?Typically, one will find E−values in a BLAST search. (BLAST is a program that finds similar protein or nucleotide sequences to your target sequence).
- The S score is a measure of the similarity of the query to the sequence shown.
- The E−value is a measure of the reliability of the S score.
The definition of the E−value is: The probability due to chance, that there is another alignment with a similarity greater than the given S score.
ماشین بردار پشتیبانی
از ویکیپدیا، دانشنامهٔ آزاد
ماشین بردار پشتیبانی (Support vector machines - SVMs) یکی از روشهای یادگیری باناظر است که از آن برای طبقهبندی و رگرسیون استفاده میکنند.
این روش از جملهٔ روشهای نسبتاً جدیدی است که در سالهای اخیر کارایی خوبی نسبت به روشهای قدیمیتر برای طبقهبندی از جمله شبکههای عصبی پرسپترون نشان داده است. مبنای کاری دستهبندی کنندة SVM دستهبندی خطی دادهها است و در تقسیم خطی دادهها سعی میکنیم خطی را انتخاب کنیم که حاشیه اطمینان بیشتری داشته باشد. حل معادلة پیدا کردن خط بهینه برای دادهها به وسیله روشهای QP که روشهای شناخته شدهای در حل مسائل محدودیتدار هستند صورت میگیرد. قبل از تقسیمِ خطی برای اینکه ماشین بتواند دادههای با پیچیدگی بالا را دستهبندی کند دادهها را به وسیلهٔ تابعِ phi به فضای با ابعاد خیلی بالاتر میبریم. برای اینکه بتوانیم مساله ابعاد خیلی بالا را با استفاده از این روشها حل کنیم از قضیه دوگانی لاگرانژ برای تبدیلِ مسالهٔ مینیممسازی مورد نظر به فرم دوگانی آن که در آن به جای تابع پیچیدهٔ phi که ما را به فضایی با ابعاد بالا میبرد، تابعِ سادهتری به نامِ تابع هسته که ضرب برداری تابع phi است ظاهر میشود استفاده میکنیم. از توابع هسته مختلفی از جمله هستههای نمایی، چندجملهای و سیگموید میتوان استفاده نمود.
تحلیل تفکیک خطی
از ویکیپدیا، دانشنامهٔ آزاد
تحلیل تفکیک خطی (Linear Discriminant Analysis -LDA) و تفکیک خطی فیشر روشهای آماری هستند که از جمله در یادگیری ماشین برای پیدا کردن ترکیب خطی خصوصیاتی که به بهترین صورت دو یا چند کلاس از اشیا را از هم جدا میکند، استفاده میشود.
شبکههای بیزی
از ویکیپدیا، دانشنامهٔ آزاد
یک "شبکهٔ بیزی" یا "شبکه باور" یا "شبکه باور بیزی" یک گراف جهتدار غیرمدور است که مجموعه ای از متغیرهای تصادفی و نحوه ارتباط مستقل آنها را نشان می دهد. به عنوان نمونه یک شبکه بیزی می تواند نشان دهنده ارتباط بین علت بیماریها با خود آنها باشد. پس با داشتن عوامل بتوان احتمال یک بیماری خاص را در یک مریض تشخیص داد.
شبکه عصبی مصنوعی
از ویکیپدیا، دانشنامهٔ آزاد
شبکههای عصبی مصنوعی (Artificial Neural Network - ANN) یا به زبان سادهتر شبکههای عصبی سیستمها و روشهای محاسباتی نوینی هستند برای یادگیری ماشینی، نمایش دانش، و در انتها اعمال دانش به دست آمده در جهت بیشبینی پاسخهای خروجی از سامانههای پیچیده. ایدهٔ اصلی این گونه شبکهها (تا حدودی) الهامگرفته از شیوهٔ کارکرد سیستم عصبی زیستی، برای پردازش دادهها، و اطلاعات به منظور یادگیری و ایجاد دانش قرار دارد. عنصر کلیدی این ایده، ایجاد ساختارهایی جدید برای سامانهٔ پردازش اطلاعات است. این سیستم از شمار زیادی عناصر پردازشی فوق العاده بهمپیوسته با نام نورون تشکیل شده که برای حل یک مسأله با هم هماهنگ عمل میکند.
با استفاده از دانش برنامهنویسی رایانه میتوان ساختار دادهای طراحی کرد که همانند یک نرون عمل نماید. سپس با ایجاد شبکهای از این نورونهای مصنوعی به هم پیوسته، ایجاد یک الگوریتم آموزشی برای شبکه و اعمال این الگوریتم به شبکه آن را آموزش داد.
این شبکهها برای تخمین (Estimation) و تقریب (Approximation)کارایی بسیار بالایی از خود نشان داده اند. گستره کاربرد این مدلهای ریاضی بر گرفته از عملکرد مغز انسان، بسیار وسیع میباشد که به عنوان چند نمونه کوچک می توان استفاده از این ابزار ریاضی در پردازش سیگنالهای بیولوییکی، مخابراتی و الکترونیکی تا کمک در نجوم و فضا نوردی را نام برد.
خلاصه اینجوری
---
ضمنا منحل شدن دانشگاه ایران رو هر چند عملی نیست، به عموم دانشگاهیان تسلیت میگم و امیدوارم که به خاطرات نپیونده...
فکر کنم چند وقت دیگه وبلاگمون به خاطر زدن پستهای یک کم علمی فیل.تر شه...
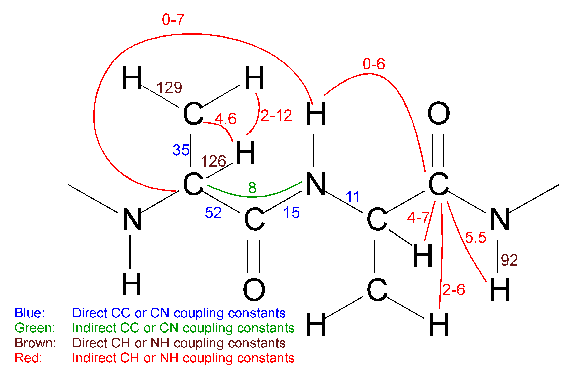
Analysis is based round the concept of a Resonance - it makes sense, but is difficult to explain. But I will try. Each resonance is associated with one particular atom in your molecule. In theory you might think that it is associated with the particular chemical shift of that atom, but the reason for having the concept of a resonance, is that the chemical shift itself is dependent on many outside influences, such as temperature, pH, salt, ligand etc. This means that any one atom is not associated with any one particular chemical shift. Thus the resonance is there to hold it all together. The reason why you can't simply use the atom as your central object is that to start with you obviously don't know which peak corresponds to which atom.
When you pick a peak in Analysis the first thing you have to do to it, is create a new resonance for each dimension. At a later stage you may then add further attributes such as the atom type (Cα or Cβ), or the full assignment, i.e. exactly which atom in the molecule it corresponds to (Asp10Cα or Trp54N). Resonances are numbered from one upwards and are always shown in square brackets in the peak labels and can be looked at and manipulated using the Browse Resonances pop-up which can be called up from the Assignment pull-down menu. The Browse Resonances pop-up is also useful for navigation purposes, as you can mark selected resonances and go to their position within a specified window.
A spin system basically contains all resonances which belong to one amino acid in a protein (or one nucleotide in DNA/RNA or one sugar ring in a carbohydrate). When two resonances are known to be in the same spin system they can be added to a new spin system. Spin systems are initially numbered one upwards and their numbers are shown in curly brackets in the peak labels. Spin systems can be manipulated in the Spin Systems pop-up which is accessed via the Assignment menu. It is possible to associate a spin system with a particular amino acid type and also to merge them (e.g. if resonances originally associated with separate spin systems later turn out to belong to the same one).






















 و یک نقطه در
و یک نقطه در  نوشته میشود, و کتِ پسی خوانده میشود. (هر متغیر دیگری میتواند جایگزین علامت پسی شود) هر کت میتواند با تعدادی بردار یکه مشخص شود,
نوشته میشود, و کتِ پسی خوانده میشود. (هر متغیر دیگری میتواند جایگزین علامت پسی شود) هر کت میتواند با تعدادی بردار یکه مشخص شود,

 نوشته میشود. مثلا برای مطابق با کت بالا عبارت است از:
نوشته میشود. مثلا برای مطابق با کت بالا عبارت است از:
 است که به صورت زیر تعریف می شود:
است که به صورت زیر تعریف می شود:
 که برای همه "کت"ها داریم
که برای همه "کت"ها داریم 








 است که
است که
{kind=link}
{kind=link}
{kind=link}
{kind=link}